
RG-1 Regulatory guidance:
Chapter 2 - Data requirements for single ingredient approval and feed registration
2.7 Guidelines for the assessment of novel feeds: microbial sources
This page is part of the Guidance Document Repository (GDR).
Looking for related documents?
Search for related documents in the Guidance Document Repository
1 Introduction
1.1 Scope
These guidelines (Section 2.7 of RG-1, Regulatory Guidance: Feed Registration Procedures and Labelling Standards) apply to novel feeds composed of microorganisms, or products derived from them, which have not previously been authorized by the Canadian Food Inspection Agency (CFIA) for use as livestock feed in Canada and/or have a novel trait.
Prior to the release of a novel feed (i.e., the manufacture, sale or import or use as livestock feed), authorization from the Animal Feed Division (AFD) of the CFIA is required.
This section of the RG-1 provides guidance for the preparation of an application for the authorization of the release of a novel microbial feed. It includes criteria that will be considered in the assessment of the safety and efficacy of a novel feed. It is not intended to explicitly define all of the data that might be required in the course of the assessment.
Learn more about novel feeds, application procedures and AFD activities
Please be aware that the information provided in these guidelines is not exhaustive and will be updated as appropriate to reflect current scientific knowledge. For further clarification, applicants are strongly encouraged to consult with the AFD. For all purposes of interpreting and applying the law, applicants are invited to consult the official version of the relevant Acts and Regulations.
1.2 Microbial feed products
Microbial products commonly used in livestock feeds are classified into one of two categories:
- viable microbial products or VMPs (bacteria, yeasts, moulds), including silage inoculants which aid in the preservation of forages by fermentation, and direct-fed microbials (DFMs) for the improvement of animal performance. Silage inoculants and DFMs may be composed of one or more strains of microorganisms. The safety and viability of each individual strain in a mixed product must be evaluated and each strain must be registered before a product can be evaluated and registered. Additional information about silage inoculants is available in Section 3.9, Registration Requirements for Forage Additives, and more information about DFMs is available in Section 3.22, Viable Microbials and Yeasts
- non-viable microbial products, including products derived from viable microbials, such as enzymes, amino acids, vitamins, and flavouring ingredients, and by-products and residuals of fermentation processes, such as spent grains and yeast biomass remaining after the production of beer or the manufacture of ethanol. Additional information about products derived from microbials is available in Section 3.6, Registration Requirements for Enzyme Supplements and Mixed Feeds Containing Enzyme Supplements, More information about fermentation by-products and residuals is available in RG-6, Regulatory Guidance: Ethanol Distillers' Grains for Livestock Feed
1.3 Classification of viable microbial products (feeds, drugs, or veterinary biologics)
A viable microbial product or products thereof for direct livestock consumption is classified as a feed, a drug, or a veterinary biologic, based on the product label claims. The onus is on the company producing the product to determine, and support, the appropriate classification of its product in a satisfactory manner.
When product label claims, company advertising claims, or scientific literature indicate that the viable microbial feed product is a drug or a veterinary biologic, applicants are advised to consult with the appropriate government body (i.e., the Veterinary Drugs Directorate at Health Canada, or the Canadian Centre for Veterinary Biologics at the CFIA). For more information about the classification of viable microbial products, including the types of claims associated with each classification and how to contact the appropriate organization, please consult Section 3.22, Viable Microbials and Yeasts.
More information about the classification of drugs vs. feeds is also available in the Guidance document on classification of veterinary drugs and livestock feeds, available on the Health Canada website.
1.4 Regulation of novel feeds
The AFD evaluates and regulates all feed ingredients, including novel feeds, in the same manner. Any feed ingredient that is new (i.e., is not already listed in Schedules IV or V of the Feeds Regulations), or has been modified such that it differs significantly from a conventional ingredient, is required to undergo a pre-market assessment and approval. The purpose of all feed assessments is the same: to ensure that the feed ingredient is safe (in terms of animal health, human health via residues in food and worker/by-stander exposure, and the environment) and effective for its intended purpose prior to marketing. The evaluation also ensures that the feed is accurately defined in the Feeds Regulations and is labelled appropriately for its safe, effective use and for consumer protection.
1.5 Research with novel microbial feed products
If novel microorganisms, or parts or products derived from them, are not ready for commercialization but will be used in livestock feeding trials or in pilot scale feed development research trials, the AFD must be notified and an application for Authorization for the Release of Novel Feeds for Research Purposes must be submitted. Please refer to RG-1, Chapter 5 – Research with Livestock Feeds for more information.
1.6 Assessment principles
For general information about how applications for registration for feed products are assessed, please refer to Chapter 1 of the RG-1.
The AFD conducts novel livestock feed safety assessments based on familiarity and substantial equivalence. Familiarity and substantial equivalence are two key concepts developed by the OECD (1993) and further elaborated by the FAO/WHO (2000).
Familiarity can be defined as the knowledge of the properties and characteristics of a microbial product and its uses and applications in livestock feeds in Canada, i.e., those listed in Schedules IV or V of the Feed Regulations. Substantial equivalence is based on the comparison of a novel product to an appropriate counterpart, taking into consideration both intended and unintended effects. A Degree of Equivalence can be established for novel microbial products if the intended uses, composition and functional properties of the product are substantially equivalent and the production strains are substantially equivalent in terms of safety to animal health, human health, and the environment. Once familiarity and a degree of equivalence are established, the safety assessment process focuses on the differences between the novel product and its counterpart and their impact on the safety of the novel product.
Essential considerations in the assessment of novel feeds from microbial sources include the safety of the production microorganism and microbial products to humans, animals and the environment. Some areas of consideration during the assessment include:
- the potential impact of horizontal gene transfer to microorganisms in the gut and in the environment
- interactions with the gastrointestinal (GI) microflora
- persistence in the gut
- potential impact on humans and the environment through shedding of viable microorganisms in livestock faecal material
- the ability of viable microorganisms to survive in the GI tract, which may impact the quality of the meat derived from the animal consuming the feed
Where microorganisms may contaminate meat, Health Canada may require an assessment of the safety of the product as a novel food.
1.7 The application process
General administrative information regarding the procedures for application for feed registration can be found in RG-1, Chapter 1, Administrative Requirements for Registration and Approval of Livestock Feed, Specific requirements are found in Part 2 below, and in the attached checklist.
Certain microbial products or ingredients have specific data requirements necessary for supporting safety or efficacy claims. Please refer to the appropriate guidance documents for additional information:
2 Characterization of the microbial product
The submission package must include a summary to provide the reader with an overview of the product, its purpose, its applications and effects on the target species, the identity of the production organism, and, where applicable, the identity of the donor micro-organism and the modification method(s).
All experimental work must be conducted in accordance with sound scientific principles and Good Laboratory Practices. The methods used to generate the experimental data must be submitted, or referenced if published.
All data submitted in support of the safety assessment, including tables, maps, figures, graphs, analytical gels and blots, should be of high quality, comparable to that accepted by reputable peer-reviewed scientific journals. Particular attention should be paid to the sensitivity and specificity of analytical methods. Appropriate controls and reference material used in the analyses and data demonstrating the reproducibility, sensitivity or precision of a method must be submitted. Statistical analyses must be conducted using appropriate techniques and raw data must be made available upon request. Literature reviews submitted in support of the safety of a product must include all references cited in the text. Where data from published literature is cited in support of the safety of the product, a copy of the publication or a journal Web site for the full text article must be provided.
Reviewers' checklists (see Appendix II of this document) provide guidance to applicants in the preparation of quality data. It is recommended that applicants review the appropriate checklist(s) for analytical techniques while assembling a data package in support of a submission.
The manufacturing process must ensure the maintenance of a pure and genetically stable strain, and the production of a consistent product, free from contamination.
The toxicological and physicochemical properties of the microbial product must be well-characterized, since the microbial preparation may range from highly purified compounds to crude fermentation product, containing media residues, cellular components and various products and by-products of microbial metabolism.
Oral toxicity studies and genotoxicity tests may be required, to confirm the absence of harmful substances that may be produced as co-products during the fermentation process or may be present as contaminants.
When the feed microbial product consists of, or is derived from, a genetically modified microorganism, additional considerations would include: the safety of the donor organism (where applicable); the characterization of the genetic modification and the novel trait; the properties of the modified organism; any unintended effects and/or secondary effects; and the disruption or alteration of metabolic pathways induced by the genetic modification. These alterations may be beneficial to the feed, may have no effects, or may introduce toxic or allergenic compounds and changes in safety characteristics of the feed. For a live microorganism, the properties conferred as a result of the genetic modification must be evaluated, and the effect of these properties on the survival, competitiveness and survival fitness of the microorganism in the GI tract and in the environment must be determined.
2.1 Product description and use
The following lists the criteria that are considered in the assessment process, and is intended to provide guidance for the preparation of an application for authorization. Please submit all applicable information:
- The identity of the product and a description, including:
- proposed name(s)
- type of product and identity of the active ingredient
- product formula, if consisting of a mixture, listing the ingredients and their proportion by weight
- physical state of the product, including any processes such as granulation or encapsulation
- purpose of the active ingredient/product
- mode of action of the active ingredient
- taxonomic identity of the production microorganism, including strain number
- Unit amount/package size
- Product label
- Directions for use, including:
- mixing information, if used with other products
- suggested rates, timing, intervals of feeding
- identification of species of intended use
- distribution, storage and handling information
- strategies for reuse, resale and disposal of unused product
- Notifications and/or regulatory approvals of other government agencies and international agencies
- A list of all ingredients, including those used in manufacturing, and a description of their mode of action or purpose
- Material Safety Data Sheets (MSDS) for the product and the ingredients
- A detailed description of the manufacturing process, including:
- a flow chart of the manufacturing process, from strain revival to product packaging, describing the steps in the preparation of the microbial product
- the composition of the growth media, from the initial inoculation steps to the production level bio-reactor
- the fermentation conditions
- a detailed description of the separation and purification steps, including all processing aids that are used during the manufacturing process
- the batch-to-batch variation in the concentration of the active ingredient and the impurities
- quality control/quality assurance procedures and protocols for purity of the production microorganism prior to and during manufacturing, production parameters, contamination, consistency and conformity of the final product, and the methods used to monitor genetic drift of the production organism
- methods for treatment and/or disposal of by-products and waste products resulting from the fermentation process. Data supporting the efficacy of the treatment and/or disposal method should be provided. This requirement is specific to microbial fermentations conducted in Canada
- a statement confirming the application of Canadian Biosafety Guidelines for lab use, and National Institutes of Health (NIH)/Good Large Scale Practices (GLSP), for large scale manufacturing, where applicable. A description should be provided on how the facility meets every point described in CBSG or NIH to ensure adequate containment
- The composition of the final product, including:
- the proportion and concentration of ingredients used to formulate the final product, including the amount of source material derived from the fermentation process, (example, 90% rice hulls (carrier), 10% fermentate)
- the percentage purity of the source material derived from the fermentation process, example, fermentate is composed of 70% active ingredient and 30% media residue. The purity of other ingredients in the product must also be provided
- the amount of active ingredient in the final product in ppm (parts per million) or per cent, or in colony forming units (CFU)/g or viable cells/g (depending on the enumeration method) when the active ingredient is a live organism. When the product is composed of a mixture of microorganisms, each strain must be described separately, and the numbers of each strain in the product must be provided
- identification of any extraneous material likely to occur in the final product
- Certificates of Analysis for three production batches or pilot scale batches for the active ingredient, demonstrating identity and concentration. Appropriate controls or standards must be included, if applicable. The methods of analysis must be described, or referenced if published. As an example, for an amino acid, the data should demonstrate the concentration and identity of the amino acid as compared to a mixture of amino acid standards
- Data supporting the presence or absence, or viability or non-viability, of the production strain in the final product, depending on the nature of the product and the specifications
- Data supporting a shelf life of at least 12 months from the date of manufacture, as shown for a minimum of three production batches of the product
- A list of all heavy metal, chemical and biological contaminants, either inherently present or introduced via the process, including:
- certificates of Analysis for biological contaminants in three production batches of the product for total plate counts, pseudomonads, Salmonella, Staphylococcus, E. coli, coliforms, yeasts and moulds (with no Aspergillus flavus or Fusarium spp. detected)
- certificates of Analysis for heavy metals, chemicals and for mycotoxins where applicable
- analytical methodologies used to quantify contaminants
2.2 Characterization of the production microorganism
The following information must be provided:
- Taxonomic classification based on a recognized nomenclature organization or refereed scientific publications, to allow for the complete identification of the microorganism, including the following: genus, species, subspecies, strain and/or type, as well as any substantiated changes in classification, and internal identification numbers if applicable. Information on the methods used to substantiate the identification along with all test data should be provided (details explained in paragraph D below). A Certificate of Analysis should be included, confirming the identification of the microorganism from three recent production batches
- Common name(s), including abbreviations or acronyms, as well as names used in other countries
- Accession numbers or other information from a recognized culture repository, if available, and a copy of the deposit certificate for the organism (and/or for the parental strain if the production organism has been genetically modified)
- Criteria, methodology and analytical data to substantiate identification and taxonomic assignation of the microorganism, including phenotypic traits (morphology, substrate usage, fermentation products, etc.) and phylogenetic analysis (16S or 23S ribosomal RNA sequencing, or other molecular taxonomic methods). Immunological tests (example, radioimmunoassay [RIA]) may be necessary depending on the source of the microorganism. For pure cultures, the information submitted should be sufficient to distinguish the microorganism from related microorganisms, and to describe its relationship to pathogens. For mixed cultures, it is necessary to provide the identification and characterization information for each strain present in the mixture along with the methods and criteria used to select and enumerate each strain. In exceptional circumstances, complex mixtures may be considered on a case-by-case basis
- The strain's origin and following history from point of isolation to acquisition (example: original habitat, environmental isolate, certificate of origin, clinical isolate, culture collection) and development (example: selection procedures, culture maintenance)
- Reports and documentation on any history of use or hazards of the strain in agriculture, food production and other industrial practices
- Other documented uses of the strain and closely related strains or species and information on exposure routes
- Any reports of disease, opportunistic pathogenicity, virulence factors, and/or toxin production associated with the strain or closely related species. Where related strains or species of microorganisms are capable of producing adverse effects, the absence of such effects should be demonstrated in the production organism; this may include data to demonstrate that genes responsible for the adverse effects in related species have been identified and their absence or suppression in the production strain has been shown
- Detailed description of any inherent antimicrobial activity
- Detailed description of any inherent antimicrobial resistance factors present in the strain and, when necessary, the characterization of the genetic determinants of the antimicrobial resistance, example, whether they are associated with mobile genetic elements or integrated in a stable manner into the chromosome. Scientific rationale, with supporting evidence, is to be provided on the safety concerns, or lack thereof, of resistance elements present in the strain
If any antimicrobial resistance is detected, the potential impact on human and animal health must be described. A review of the scientific literature should be conducted to document any observed resistance in other strains of the same species for target antibiotics
A national standard should be followed (example,European Food Safety Authority) when establishing minimum inhibitory concentration (MIC) values for the notified strain. Control strains should be included with average MIC values for the species and pathogenic members of the genus
- Information on the presence of mobile genetic elements (example, insertion sequences, transposons, plasmids, prophages)
- If the organism has not been genetically modified, a statement to that effect must be submitted
2.3 Development of the modified microorganism
This subsection is for consideration only if the organism has been genetically modified by recombinant nucleic acid techniques, deletion, rearrangement or suppression of native DNA, classical mutagenesis techniques, selective pressure, or other techniques.
Sufficient information on the process used to effect the genetic modification should be submitted to enable the characterization of the modified microorganism, and permit comparison with the conventional or unmodified counterpart. The identification of all genetic material introduced, modified or deleted in the recipient organism and scientific analysis of the data supporting the molecular characterization should allow an assessment of both safety and potential secondary and unintended effects. These effects could be caused by insertional activation or inactivation of genes, transcription of vector sequences, and/or the impact of foreign gene products on the metabolism of the host, leading to altered levels of existing gene products or metabolites.
Newly expressed material, resulting from introduced genetic material or modified native material, should be fully characterized, and similarity to products from traditional sources should be described where appropriate. If novel constituents (other than those resulting from the intentional modification of genetic material) are identified, further studies are required to characterize the unintended effects and their impact on the safety of the production organism and the microbial product. It should be demonstrated that introduced changes do not cause adverse effects, changes are stable, and potential gene transfer to other species is not enhanced. Where genetic modifications alter the expression of a traditional metabolite or constituent, sufficient information on the anabolic or catabolic pathways should be provided to enable an assessment of possible secondary effects on related pathways and metabolite production.
The following information must be provided:
- Description of the Donor Organism(s) and Intermediate Organism(s) (where applicable), including:
- taxonomic names, based on the most current recognized nomenclature organization or refereed scientific publications, to allow for the complete identification of the donor organism, including any substantiated changes in nomenclature and any previous nomenclatures
- common name(s), including alternatives, abbreviations or acronyms, as well as names used in other countries
- accession numbers or other information from a recognized culture repository, if applicable, and the deposit certificate
- criteria and methodology to substantiate identification and classification
- origin of the donor organism(s) and intermediate organism(s), including history during development (example, selection procedures, culture maintenance)
- reports and documentation on any past and present uses of the donor organism, or closely related organisms in feeds, foods or other applications, and information on exposure routes, hazards, etc
- any reports of disease, pathogenicity, including opportunistic pathogenicity, virulence factors or toxin production by the donor organism or by closely related organisms
- Description and Characterization of the Genetic Modification, including:
- description of the genetic modification, including a list of the genes introduced, modified or deleted, their function and the resultant novel traits
- purpose of the modification and the intended properties conferred to or lost by the genetically modified microorganism as a result of the modification
- detailed description of the recipient strain construction/modification process, including:
- a) a flow diagram, including the names of the donor, recipient and vector DNA and the steps (linked in the order of their development) used to construct the final modified microorganism
- b) a description of the method(s) used to introduce, delete or modify the genetic material and the resultant novel trait(s), including references or citations
- c) when vectors are used during the genetic modification, the following must be provided:
- the name and source of the vector(s)
- the origin and characterization of the sequences of the vector(s), including regulatory elements (example, promoters, modifiers, enhancers, signal peptides and terminators), replicons, selective marker genes, linkers and any foreign DNA, in case the vector or DNA sequences from the vector are part of the genetically modified microorganism. Citations or references should be included on the development of the vector
- the natural pathogenicity and infectivity of the vector
- the method used to disarm the vector, such as removal of antimicrobial resistance markers or inactivation of transposons and other mobile elements, if applicable
- d) a description of the intermediate recipient organisms used to produce or process DNA prior to introduction into the final recipient organism; and
- e) a physical map of each genetic construct, detailing size and positions of all coding and non-coding sequences, all regulatory elements, origins of replication and transfer, marker genes, location of restriction sites, primers used for PCR analysis, and regions used as probes. A table identifying and referencing each component should be included with the map. Appendix III is provided as a model to follow for the submission of vector characterization data
- 2) Characterization of inserted, deleted or otherwise modified genetic material in the final recipient organism, as follows:
- a) identification of the insertion, deletion or modification sites
- b) the complete sequence of the inserted and/or modified genetic material
- c) the size, identity and copy number of the genetic material inserted at each site
- d) the organization and orientation of the modified genetic material at each insertion or modification site
- e) for all coding regions, demonstration of whether complete or partial copies were inserted into the genome, and data to show that promoters, leader sequences, terminators and other regulatory regions are inserted intact with the coding regions whose expression they are designed to regulate
- f) identification of any open reading frames (ORF) within the inserted material or created by the insertions with contiguous chromosomal or plasmid DNA, including those that could result in chimeric proteins. When transcription of such ORFs occurs, the safety of the chimeric protein and its impact on the safety of the product must be assessed
- g) marker gene systems: if the gene transfer system employs a marker gene that is expressed, information to support the safety of the marker gene system must be provided
- h) the stability of the genetic modification and the arrangement of the genetic material in the recipient organism, generated from production scale or pilot scale fermentation, for a minimum of three replicates. Gel electrophoresis images should be provided, showing stability of the modification over multiple generations
2.4 Characterization of the novel trait(s)
Provide the following information about the new modification, whether introduced or modified native material:
- a description of the morphological and physiological characteristics, growth patterns and growth rates of the modified microorganism as compared to the unmodified parental (or isogenic) strain
- data that demonstrate the genetic stability of the modified organism and that all novel traits are expressed and inherited in a stable manner
- the gene product(s), breakdown products, by-products and their metabolic pathways, and the analysis of transcripts or expression products to identify any new substance that may be produced
- the function of the gene product(s)
- the phenotypic description of the new trait
- the level and the site of expression of the gene product(s) including those of the marker gene(s), if applicable
- a determination whether gene expression is constitutive or inducible, with a description of the inducing agents
- the activity of the gene product(s), breakdown products and by-products in the host microorganism, and any resulting changes to existing metabolic pathways (including altered accumulation and storage patterns)
- for microorganisms used for the processing or treatment of feeds (example, forage inoculants, acidifiers), the activity of the gene product(s), breakdown product(s), and by-product(s) should be examined under the conditions of use
- for all coding regions inserted or modified, expressed protein characterization data, including:
- any modifications to the amino acid sequence and properties of the protein, or post-translational modifications to the expressed protein, such as glycosylation or phosphorylation, which may have occurred when the coding sequence has been modified or transferred from one organism to another
- where protein structure has been modified, data to demonstrate how the amino acid modifications affect sites critical to structure or function, and amino acid sequence homology of the novel protein to the amino acid sequences of closely related proteins from the same family and to native proteins, indicating the locations of the amino acid differences and whether they occur in variable or conserved regions
- The following data may be required for novel enzymes:
- a) chemical name, enzyme commission classification number, and Chemical Abstract Service (CAS) registry number
- b) a description of the catalytic functions and mode of action
- c) the substrate specificity for each catalytic function
- d) known co-factors required for enzyme activity
- e) pH optimum and profile
- f) temperature optimum and profile
- g) kinetic properties for each substrate assayed, including Vmax, Km, and Kcat. Where the catalytic properties of a novel enzyme differ from the properties of the native enzyme, a valid scientific interpretation must be provided, explaining the significance of this difference in activity, based on the variation in the range of activity exhibited by enzymes belonging to the same family
- h) for structurally modified enzymes, the impact of the modification on the activity of the novel enzyme, as compared to the native enzyme, at a range of pH, temperature and other characteristics that may affect the activity of the enzyme under the conditions of usage
- i) the stability and metabolic fate of the novel enzyme
3 Inactivation of antimicrobials in feed
Antimicrobials are routinely mixed into livestock feeds. A determination of the potential for their inactivation during storage in a mixed feed, as a result of inherent, acquired or incorporated antimicrobial resistance, should be conducted. Antimicrobial efficacy, in the presence of the microbial product, should be monitored over a reasonable length of time. If biochemical parameters such as pH or moisture content of the feed preclude the expression or activity of the antimicrobial resistance protein, this study may be waived.
4 Animal and human safety
Safety information must be provided to identify the type and severity of potential health risks that the microorganism(s) or product(s) may pose to humans or animals. Where no reported adverse effects are found, describe any steps taken to ascertain the lack of reports of adverse effects (example, the databases searched, the period search, the search strategy and keywords used). Required information may include but is not limited to, the following:
- Relation to any microorganism(s) associated with adverse effects
Describe the relationship of the microorganism to other infectious, toxic, pathogenic, or virulent microorganisms. If the microorganism is taxonomically related to pathogenic organisms, data must be provided demonstrating the absence of pathogenic potential in the microorganism, including the absence of genes associated with pathogenic effects - Potential adverse health effects
The ability of the novel feed to cause disease in humans and animals must be assessed. Evaluation parameters include the nature and degree of infectivity, toxicity, and pathogenicity of the novel feed. Report on any factors that may predispose an animal to the adverse effect, provide all available epidemiological data, and supply all information on occurrences of opportunistic pathogenicity - Potential for toxicity
Potential toxins produced by the microorganism(s), and any metabolites that are related to toxins produced by other microorganisms must be identified and their relevance assessed. The fate and concentration of these toxins in the final product must be determined and toxicity studies may be required. In circumstances where a genetic modification results in the production of novel substances (i.e., proteins) or the modification of existing ones, data must be submitted in support of the safety of the novel or modified substance(s). Potential toxicity of novel proteinaceous substances can be evaluated by comparing the amino acid sequences of the novel protein with known toxins, and assessing stability to heat and to degradation in appropriate gastric and intestinal models - Potential for allergenicity
Possible allergenicity of novel proteins to animals and to humans must be evaluated. Currently, there are no reliable animal models or definitive tests to predict allergenicity. Applicants are advised to follow the stepwise case-by-case approach elaborated by the FAO/WHO and endorsed by the Codex Alimentarius. For guidance, refer to the Codex Alimentarius guideline for the conduct of food safety assessment of foods produced using recombinant-DNA microorganisms.Factors to consider in the assessment of potential allergenicity include the production organism, the donor organism, the degree of amino acid similarity between a novel protein and known allergens, susceptibility of the novel protein to degradation, its heat and acid resistance, and its reactivity with blood serum IgE of sensitive individuals
- Potential for gene transfer
Transfer of DNA by transmissible genetic elements (example, plasmids or conjugative transposons) to microorganisms of the gastrointestinal tract must be evaluated. For microbial products such as enzymes, amino acids, and vitamins, the presence of genetic material in the final product must be assessed and the significance of that material should be evaluated - Potential for persistence in the gastrointestinal tract
For live microbial products, the ability of the microorganism to survive passage through the gastrointestinal (GI) tract, its persistence in the GI tract, and its potential effect on the microflora of the GI tract should be assessed - Animal Studies
Laboratory animal studies and/or livestock feeding trials may be required to address any toxicological concerns arising from an assessment of all data submitted. The following toxicological tests are recommended, as a minimum, for each novel feed from a microbial source:- a) Viable microorganisms:
- acute oral infectivity and toxicity
- acute pulmonary infectivity and toxicity
- acute dermal toxicity
- mutagenicity (example, genotoxicity, point mutation, chromosomal aberrations, DNA repair, with and without metabolic activation)
- short-term toxicity (i.e., 30- to 90-day trials)
- b) For fermentation products:
- acute oral infectivity and toxicity
- acute dermal toxicity
- mutagenicity (example, genotoxicity, point mutation, chromosomal aberrations, DNA repair, with and without metabolic activation)
- short-term toxicity (example, 90-day trials)
Other tests may be required based on the assessment of these studies. Appropriate scientific rationale should be provided for the omission of any studies. See Appendix 1 for a more detailed description of all studies.
- a) Viable microorganisms:
- Potential exposure to humans
Studies may be required estimating the potential exposures to humans. Exposure must include the microorganism(s), the gene product(s), the breakdown product(s), etc. These should provide information on the level of exposure, and the most likely route of exposure. Please take into account the amount of product handled by the workers, the frequency and duration of exposure, dilution factors, and the use of protective equipment which may mitigate some of the exposure parameters
5 Environmental safety
This part of the guidelines describes the requirements concerning biological and ecological characterization of the novel feed. The information should provide sufficient data to evaluate the potential environmental effects, including the environmental fate of the microorganism(s), its metabolites, breakdown product(s), and any by-product(s). The goal is to identify and assess the extent of any potential hazards, i.e., the short and long-term presence of the novel feed in the environment. If factors that control the survival and multiplication of the microorganism are known, it is easier to predict that organism's survival and multiplication under its condition of use. Where no reported adverse effects are found, please provide reasons for potential false-negatives or a lack of information, including a lack of published research, limitations to the databases searched, limitations to the search parameters including timeframes, the search strategy, or the keywords used. Results must be interpreted using a valid scientific rationale. Information should include but is not limited to:
- Culture requirements and characteristics
Describe the relevant culture conditions and physiological properties affecting persistence, proliferation, and growth of the microorganism. These may include selectable traits such as temperature, pH, substrate moisture, nutritional dependence, oxygen requirements, energy sources, susceptibility or tolerance to antimicrobial agents, disinfectants and antiseptics. The sensitivity to environmental factors (example, sunlight, moisture level) should also be considered. This section should also include a discussion on the possible reversion of the modified microorganism to the wild type - Life cycle
Describe the various forms of the microorganism during its life cycle. These are important because the various forms may differ regarding persistence, proliferation or dispersal. These may include symbiotic forms, viable but non-culturable forms, dormant stages, spore production, and so on - Geographical distribution and level of occurrence
Describe other habitats in Canada where the microorganism is found. The indigenous levels of the microorganism in those habitats must be discussed, and the displacement of microbial community structure or function should be considered. In the specific case of genetically modified microorganisms, any new habitats that would favour the proliferation or dispersal of the modified microorganism must be identified - Host range
Describe host specificity. Any other non-target species (represented by plants, invertebrates or vertebrates, depending on the purpose of the product) that may be affected by the novel feed must be examined - Mode of action
Describe how the microorganism exerts its effect on the target species, if known - Potential adverse health effects
The ability of the novel feed to cause adverse effects on the health of non-target plants, animals and microorganisms must be assessed. Examples of suitable non-target indicator species can be found in Environment Canada or United States Environmental Protection Agency guidelines. Parameters will include the nature and degree of infectivity, toxicity, and pathogenicity of the novel feed. Report on any predisposing factors related to the effect, and on any available epidemiological data - Toxin production
Any potential toxins produced by the novel feed and any metabolites that are related to toxins produced by other microorganisms must be identified, and their relevance must be assessed. Their fate and concentration in the final product must be determined - Potential for gene transfer
The transfer of DNA by plasmids or conjugative transposons to the environment (i.e., to soil microorganisms, wildlife, etc.), must be assessed - Biological diversity
The effect on biological diversity (example, distance to populated or protected areas; identification of, and impact on, endangered or threatened species; or any biological resources of social or economic importance in the area of intended use) must be assessed
6 Regulatory decision
Based on the information received, and after evaluating the potential impact on livestock, humans and the environment, the AFD will:
- authorize the release of the novel feed where there is minimal risk (conditions may be imposed on authorizations to manage risk)
- refuse to authorize the release of the novel feed where its use poses an unacceptable risk
- follow-up with the applicant (by sending what is known as a "deficiency letter") for additional information that the AFD requires to complete the assessment. The AFD may consider the application closed if no response to the deficiency letter is received after 60 days from the date the deficiency letter is sent
Once a novel feed (including those with a novel trait) has received authorization (i.e., it gets listed in Schedule IV or V or is determined to be substantially equivalent to an ingredient already listed in the Schedules), it is no longer considered to be novel
7 New information requirements
If at any time the proponent becomes aware of any new information regarding risk to the environment, to livestock, or to human health that could result from the release of the novel feed, they must immediately provide this information to the CFIA. On the basis of any new information, regardless of the source, the CFIA will re-evaluate the potential impact on and risk to the environment, including the potential impact on and risk to human or animal health, with respect to the livestock feed use. The CFIA may impose additional conditions respecting the release of the novel feed or its use as livestock feed, change the conditions respecting these authorizations, or cancel the authorizations and require the proponent to stop the release of the novel feed or its use as livestock feed and take any appropriate action necessary to eliminate or minimize the risk.
These Guidelines for the Assessment of Novel Feeds: Microbial Sources, have been prepared to provide guidance regarding the submission of an application for the authorization of the release of a novel feed, as may be required under the Feeds Regulations. Please be aware that the information provided in these guidelines is not exhaustive and will be updated as appropriate to reflect current scientific knowledge. For further clarification, applicants are strongly recommended to consult with the CFIA. For all purposes of interpreting and applying the law, applicants are invited to consult the official versions of the relevant Acts and Regulations.
8 Definitions
- Biotechnology:
- The application of science and engineering in the direct or indirect use of living organisms or parts or products of living organisms in their natural or modified forms (Canadian Environmental Protection Act).
- Carrier DNA:
- DNA used to expedite the preparation or the introduction of genetic material into a microorganism but which is itself not part of the construct.
- Codex Alimentarius:
- (Latin for Food Law): International organization created in 1963 by the UN's Food and Agriculture Organization (FAO) and World Health Organization (WHO) to develop food standards, guidelines and related texts, such as codes of practice, under the Joint FAO/WHO Food Standards Program. The main purposes of this Program are protecting the health of consumers and ensuring fair trade practices in the food trade, and promoting coordination of all food standards work undertaken by international governmental and non‑governmental organizations (Codex alimentarius)
- Coding region:
- A DNA sequence which acts as the template on which mRNA is synthesized during transcription – the sequence can be translated to produce a protein. This region is composed of the coding strand (the sense strand, or the plus (+) strand) whose sequence is complementary to that of mRNA, and the non-coding strand (complementary to the coding strand) which is also called the antisense strand or the minus (-) strand.
- Construct:
- An engineered DNA fragment (example, a plasmid) which contains, but is not limited to, the DNA sequences to be integrated into a target microorganism's genome.
- Counterpart:
- A microbial product selected by the applicant to which the novel product will be compared. The chosen counterpart should be, if possible, the closest genetic equivalent and may be a previously assessed and authorized novel feed. If the closest genetic equivalent is not similar to the novel feed, consideration may also be given to comparison with those products the novel feed was designed to emulate.
- Database citations:
- Publically accessible sources of nucleotide or protein sequence information. Five commonly used databases and their website addresses are:
- GenBank:
- An annotated collection of all publicly available DNA sequences maintained by the National Institute of Health (NIH).
- DNA Data Bank of Japan:
- The officially certified DNA bank of Japan, which collects DNA sequences from researchers.
- EMBL Nucleotide Sequence:
- A database of DNA and RNA sequences collected from the scientific literature, patent applications, and directly submitted from researchers and sequencing groups.
- UniProtKB/Swiss‑Prot Protein Knowledgebase:
- A database of protein sequences produced collaboratively by Amos Bairoch (University of Geneva) and the EBI.
- FARRP Allergen Protein Database:
- A database containing a list of unique known and putative allergens that were identified by searching publicly available protein databases. This database is managed by a panel of scientists and clinicians who are actively involved in reviewing data for inclusion of proteins in the database by comparing peer reviewed publications supporting the classification of the proteins as allergens or putative allergens following predetermined guidelines.
- Drug:
Includes any substance or mixture of substances manufactured, sold or represented for use in
- the diagnosis, treatment, mitigation or prevention of a disease, disorder, abnormal physical state, or its symptoms, in human beings or animals
- restoring, correcting or modifying organic functions in human beings or animals
- disinfection in premises in which food is manufactured, prepared or kept (Food and Drugs Act)
A nutritionally-enhanced feed is considered a feed when it is for the purpose of providing nutritional requirements or preventing or correcting nutritional disorders. A nutraceutical or "functional feed" with health benefit claims may be considered a feed or a drug based on the mode of action and the nature of the active ingredient. In this case, the Feed Program assesses the registration application in the preliminary review stage and a decision is made based on the information provided by the applicant.
- Environment:
Means the components of the Earth and includes
- air, land and water
- all layers of the atmosphere
- all organic and inorganic matter and living organisms
- the interacting natural systems that include components referred to in paragraphs a to c. (Feeds Regulations)
- Familiarity:
- The knowledge of the characteristics of a feed and experience with the use of that feed in Canada.
- Feed:
Any substance or mixture of substances containing amino acids, anti-oxidants, carbohydrates, condiments, enzymes, fats, minerals, non-protein nitrogen products, proteins or vitamins, or pelletizing, colouring, foaming or flavouring agents and any other substance manufactured, sold or represented for use
- for consumption by livestock
- for providing the nutritional requirements of livestock
- for the purpose of preventing or correcting nutritional disorders of livestock, or any such substance for use in any such substance or mixture of substances (Feeds Act)
- Genetic Modification:
- Any intentional change to the heritable traits of an organism. This includes, but is not limited to, change brought about by: recombinant nucleic acid techniques, somaclonal variation, electroporation, artificially induced mutagenesis, and similar techniques.
- Genetically Modified Microorganism (GMO):
- For the purposes of this document, means bacteria, yeast and filamentous fungi in which the genetic material has been changed through a genetic modification.
- Insert:
- That part of a construct which is integrated into the recipient microorganism's genome.
- Irritant:
- Any agent capable of eliciting an abnormally excited or sensitive condition in a body part of a human or other animal.
- Livestock:
- Means horses, cattle, sheep, goats, swine, foxes, mink, fish, rabbits and poultry and includes such other creatures as may be designated by regulation as livestock for the purposes of [the Feeds Act]. (Feeds Act).
- Microorganisms:
- Commonly understood to include algae, bacteria, fungi (yeasts and moulds), protozoa and viruses.
- Noncoding region:
- DNA sequences which lie outside of an open reading frame and which are not translated to become part of a protein. These might include scaffold attachment regions, promoters, leader sequences, enhancers, introns, terminators, and any other sequences that are used for gene expression either in the microbe or other hosts, such as origins of replication, transposable elements, T-DNA borders, lox sequences, etc.
- Novel Feed:
Means a feed, comprising an organism or organisms, or parts or products thereof, that:
- is not set out in Schedule IV or V [of the Feeds Regulations]
- has a novel trait. (Feeds Regulations)
All feed ingredients approved for use in Canada are listed in Schedules IV or V of the Feeds Regulations. A feed not originating from an organism or products thereof, and not listed in Schedule IV or V of the Feeds Regulations, is considered "Novel" (example, a flocculant chemical ingredient, or tree bark powder ingredient), and must be assessed for both safety and efficacy. However, a feed is considered "New" in cases where it originates from an approved ingredient (example, a purified source of an existing ingredient). In these cases a safety assessment may not be required for ingredient approval. The Feed Program assesses whether a feed is Novel or New based on the information provided by the applicant.
- Novel Trait:
In respect of a feed, means a characteristic of the feed that
- has been intentionally selected, created or introduced into the feed through a specific genetic change
- based on valid scientific rationale, is not substantially equivalent, in terms of its specific use and safety both for the environment and for human and animal health, to any characteristic of a similar feed that is set out in Schedule IV or V. (Feeds Regulations)
- OECD Consensus Documents:
- Reports published by the Organisation for Economic Co‑operation and Development (OECD) that contain technical information for use in the regulatory assessment of products of biotechnology. These documents are mutually recognized among member countries of the OECD.
- Release:
- Means any discharge or emission into the environment of a feed or livestock product produced from the feed, or exposure of a feed or livestock product produced from the feed to the environment. (Feeds Regulations)
- Stability:
- The ability of an introduced trait to be expressed in the genetically modified microorganism through successive generations in a consistent, reliable, and predictable manner.
- Substantial Equivalence:
- Equivalence of a novel feed from a microorganism, in terms of its specific use and safety to the environment and human and animal health, to that of a feed from the same species that is in use and generally considered as safe in Canada based on valid scientific rationale.
- Trait:
- The phenotypic characteristic(s) conferred to the recipient microorganism by a genetic modification.
- Unintended Effect:
- Unintended characteristic, property or trait acquired, modified or lost by a microorganism as a result of a genetic modification. Unintended effects may be beneficial, neutral or deleterious with respect to the properties of a microorganism and its safety to livestock, humans and the environment.
- Vector:
- An autonomously replicating DNA molecule into which foreign DNA is inserted and then propagated in a host cell.
- Veterinary Biologic:
Means
- a helminth, protozoa or microorganism
- a substance or mixture of substances derived from animals, helminths, protozoa or microorganisms
- a substance of synthetic origin that is manufactured, sold or represented for use in restoring, correcting or modifying organic functions in animals or for use in the diagnosis, treatment, mitigation or prevention of a disease, disorder, abnormal physical state, or the symptoms thereof, in animals. (Health of Animals Act)
Veterinary biologics include vaccines, bacterins, bacterin‑toxoids, immunoglobulin products, diagnostics kits, and any veterinary biologic derived through biotechnology.
Appendix I: Description of toxicity/infectivity/pathogenicity tests
The purpose of these tests is to assess the capability of novel microbial feeds to elicit toxicity and to cause disease and tissue injury, by invasion of, and multiplication within, animal or human tissues or the environment.
1. Toxicity tests
The type of toxicity data generally considered is outlined below. Please be advised that appropriate positive and negative controls are required for assays when such controls are applicable to the test. The best way to ensure the tests are conducted appropriately, and using the correct in vitro or in vivo model, is to ensure they are done in accordance with standard toxicity test methods as published by the U.S. Food & Drug Administration, the Organisation for Economic Co-operation and Development (OECD), or ASTM International.
- Rate and degree of absorption:
For each route by which a substance can be taken into the body (oral, dermal, respiratory, etc.), absorption tests determine how extensively and how rapidly it can be absorbed - Distribution, metabolism and elimination data:
These tests describe the fate of a chemical once it is absorbed into the body. Questions which should be answered by these data are: Where does it go? Does it accumulate? How is it broken down or transformed? By what route and how quickly is it eliminated? This information provides an indication of an organism's ability to tolerate short- or long-term exposures, either at high or low concentrations. It is useful information in designing toxicity tests (example,in dose selection). It also aids in extrapolating animal data to human conditions - Acute toxicity:
Acute exposure tests examine the effects of short-term, single exposures to high concentrations of a substance. Exposures in these tests are typically 24 hours or less, with effects being monitored for up to two weeks. These data are useful for establishing the relationship between the dose and the response, for ranking substances according to their relative acute toxicity, for classification, and for precautionary label statements. Acute toxicity data is used to obtain preliminary information on specific toxic effects of substances and how these may be produced (mode of action). Some specialized acute tests are described below:- i) Acute median lethality:
The concentration of a substance which, when administered once or for a short time to a group of animals, will cause death in half of the animals. This is expressed as the LD50 (lethal dose, in mg/kg of body weight), or the LC50 (lethal concentration, in parts per million). Suggested routes of exposure include oral, dermal, and inhalational - ii) Dermal/ocular Irritation:
This determines if a substance has the potential to cause irritation or cell death (necrosis) when in contact with the skin or eyes.Please note that dermal or ocular testing is not always required. Please contact the AFD for guidance regarding whether you need to perform this test.
- iii) Dermal sensitization:
This evaluates the potential for being able to "sensitize" organisms. Organisms may become more sensitive to a substance after an initial exposure and develop allergic reactions in subsequent exposures.Please note that dermal sensitization testing is not always required. However, if the characterization data indicate that the microorganism is closely related to a known dermatophyte, dermal infectivity testing may become necessary. Please contact the AFD for guidance regarding whether you need to perform this test.
- i) Acute median lethality:
- Mutagenicity:
These are screening tests used in determining a substance's potential for causing genetic mutations. Mutations to DNA may lead to cancer or gene-based malformations in offspring. At least two types of tests are performed, conventionally one with bacteria and one with mammalian cell cultures. It is also necessary to perform tests with and without metabolic activation, that is, to determine if interaction with metabolic processes in the body makes a substance mutagenic - Short-term toxicity:
Short-term toxicity studies involve repeated exposure to substances over a longer time frame (i.e., 30-90 days). They are useful for detecting most longer-term adverse health effects, for establishing a threshold level at which no effects are observed (NOEL), for establishing possible cumulative effects of exposure, species and other types of variation and for suggesting appropriate conditions for chronic tests, if deemed necessary. A 90-day oral study is typical, and inhalation or dermal studies may be appropriate, depending on typical exposure parameters - Carcinogenicity:
Where indicated by earlier tests or other data, a substance will be examined for its ability to induce cancer (tumours) in animals. Tests are typically conducted over a major portion of the animal's life span often combined with chronic toxicity tests - Epidemiological studies:
This type of study contains a compilation and analysis of information on humans who have been occupationally or accidentally exposed to a substance - Chemical interaction:
The toxic effect of a substance can sometimes be altered – increased, decreased, or changed entirely – by interactions with other substances. A toxin may be rendered harmless by another, or its effect may be magnified. Data to identify and measure the significance of such interactions improves the hazard assessment for the substance
2. Acute bacterial or viral infectivity
This test involves intravenous injection of a range of single doses of the active ingredient into an appropriate animal subject. This study provides information on health hazards likely to arise from a single high dose exposure when the skin is bypassed as a barrier. It is useful as an indication of the inherent infectivity of an organism. It would generally only be used for bacteria of unknown infectivity and viruses
3. Acute fungal or protozoan infectivity
This test involves intraperitoneal (into the peritoneum, i.e., the abdominal cavity) injection of a range of single doses of the active ingredient into an appropriate animal subject. This study provides information on health hazards likely to arise from a single high dose exposure when the skin is bypassed as a barrier. It is useful as an indication of the inherent infectivity of an organism. It would generally only be used for fungi and protozoa. If, based on the physical characteristics of the microorganism, testing by the intravenous route is possible, then the intravenous route should be considered for fungi and protozoa instead of the intraperitoneal route
Appendix II: Molecular data checklists
| Checklist | Included? |
|---|---|
| The Northern blots have a figure number and title. | |
| Lanes are labelled on the blot. | |
The figure legend describes each lane of the blot, including a description of the following for the RNA that was loaded:
|
|
| The text or the figure legend describes how RNA was extracted prior to Electrophoresis. | |
| The blot has appropriate positive and negative control lanes (positive control might demonstrate hybridization of the probe with itself; negative control might be the unmodified isogenic microorganism). | |
| The gel system and Northern hybridization protocol are described in the text or in a cited literature reference. Any modifications of the cited protocols are described in the submission text. | |
| The position of molecular size standards on the gel is indicated and they cover an appropriate size range for the fragments that are expected to be detected on the blot. | |
| The probe used for the hybridization is described adequately (in the text or in a figure) to enable interpretation of the results. | |
| The methodology or citation for the quantitative analysis (if done) has been provided, and a sufficient number of replicates or samples have been tested to determine whether there are significant differences between samples or treatments. | |
| Any superfluous bands or background signals are properly explained. |
| Checklist | Included? |
|---|---|
| The Southern blot has a figure number and title. | |
| The lanes are labelled on the blot. | |
The figure legend describes each lane of the blot, including the following for the DNA that was loaded on the gel:
|
|
| The gel has appropriate positive and negative control lanes (positive control might demonstrate hybridization of the probe with itself; negative control might be the unmodified isogenic parent microorganism). | |
| The gel system and Southern hybridization protocol are described in the text or in a cited literature reference, and any modifications of the cited protocols are described in the petition text. | |
| The positions of the molecular size standards are indicated, and they cover an appropriate size range for the fragments that are expected to be detected on the blot. | |
| If an entire plasmid was used as the probe for the hybridization, it must be described adequately in the text or in a figure to enable interpretation of the results. | |
| If a restriction fragment was used as the probe for the hybridization, it must be described adequately in the text or in a figure to enable one to interpret the results. | |
| Any superfluous bands or background signals are explained. |
| Checklist | Included? |
|---|---|
| The dot blots have a figure number and title. | |
| The lanes are labelled on the blots. | |
Each figure legend describes each lane of the blot, including a description of the following for the RNA that was loaded:
|
|
| The text or figure legend describes how RNA was extracted prior to blotting onto the solid support. | |
| The blot has appropriate positive and negative control lanes (positive control might demonstrate hybridization of the probe with itself; negative control might be the unmodified isogenic parent microorganism). | |
| The dot blot system and hybridization protocols are described in the text or in a cited literature reference, and any modifications of the cited protocols are described in the submitted text. | |
| The probe used for the hybridization is adequately described (in the text or in a figure) as to enable interpretation of the results. | |
| The methodology or citation for quantitative analysis (if done) has been provided and a sufficient number of replicates or samples have been tested to determine whether there are significant differences between samples or treatments. |
| Checklist | Included? |
|---|---|
| Figure number and title are included. | |
| Lanes are clearly labelled on the blot. | |
The figure legend describes each lane of the blot, including the following for the protein loaded:
|
|
| The protein extraction method is adequately described in the text or in the figure. | |
| The antibody or antiserum preparation protocol is adequately described in the text, including an adequate description of the antigen and its purity. The specificity of the antibody or antiserum has been determined and is described in the text or in a cited literature reference. | |
| The gel system and the blotting protocol are adequately described in the text or in a cited literature reference. | |
| The position of the molecular weight standards is indicated, and the standards cover the appropriate range for the proteins expected to be detected on the blot. | |
| The blot includes appropriate positive and negative controls. | |
| A normal serum control was conducted. | |
| Superfluous bands and background signals are properly explained. | |
| The methodology or citation for the quantitative analysis (if done) has been provided, and a sufficient number of replicates or samples have been tested to determine whether there are significant differences between samples or treatments. |
| Checklist | Included? |
|---|---|
| The PCR gel has a figure number and title. | |
| The lanes are labelled on the gel. | |
The figure legend describes each lane of the gel, including a description of the following for the DNA that was loaded:
|
|
| The position of the molecular weight standards is indicated and covers an appropriate size range for the fragments that are expected to be detected on the gel. | |
| The text or figure legend describes the PCR amplification method performed prior to electrophoresis. | |
| The primers used for amplification are described in the text or in the figure sufficiently, to enable interpretation of the results. | |
| The gel has appropriate positive and negative control lanes (positive control might demonstrate specificity of the primers and the ability to amplify the appropriate size band; negative controls might include amplification with DNA from the unmodified isogenic microorganism and amplification in absence of DNA template). | |
| The plasmid or restriction fragment used as the positive control template, is clearly described in the text or in the figure legend, so as to enable interpretation of the PCR results. | |
| The gel system and the PCR protocol are described in the text or in a cited literature reference, and modifications to the cited protocol (if applicable) are described in the text. |
| Checklist | Included? |
|---|---|
| The table has a number and title | |
| All entries are clearly identified in the table and are described in the text or table legend. | |
| The sample preparation method is described. | |
| The antibody or antiserum preparation protocol is adequately described in the text, as well as the antigen and its purity. The specificity of the antibody or antiserum has been demonstrated and described in the text or in a cited literature reference. | |
| The ELISA protocol used is described in the text or cited in the scientific literature, and any modifications to a cited protocol are described in the text. | |
| The positive controls (example,purified protein) and negative controls (example, normal or preimmune serum, non-transformed material) were used. | |
When ELISA is being used to quantify protein expression in genetically modified microorganisms:
|
| Checklist | Included? |
|---|---|
| The figure (or table) has a title and a legend. | |
| For graphical representations or tables, the axes or columns are labelled and the units indicated. | |
| The scale of the figure accurately represents the data and allows interpretation of the data. | |
The legend or text describes:
|
|
| The text or legend describes the extraction and purification of the enzyme and the degree of purification achieved. | |
| The assay method and relevant information concerning the enzyme have been provided in the text or in a cited literature reference. | |
| Appropriate controls are included in the assay. | |
| The stability of the enzyme has been taken into account in the design of the assay or the interpretation of the data. | |
| The kinetics of the enzyme have been calculated and, where possible, compared to published data. | |
| If quantitative analysis was performed, a sufficient number of replicates or samples have been tested to determine whether there are significant differences between samples or treatments, and statistical analysis was performed. |
Appendix III: Vector description
This information is taken from the document Molecular Genetic Characterization Data, prepared jointly by the CFIA, Health Canada and USDA-APHIS in 1998. It describes the critical elements to be considered during the review of the molecular genetic characterization data of transgenic plants. The same format should be followed for the submission of molecular characterization data of microbial products.
| Genetic Element |
SizeTable Note 1 Kb |
Function and Source |
|---|---|---|
| RB | 0.36 | A restriction fragment from the pTiT37 plasmid containing the 24 bp nopaline-type T-DNA right border used to initiate the T-DNA transfer from Agrobacterium tumefaciens to the plant genome (Depicker et al., 1982) |
| E35S | 0.62 | The cauliflower mosaic virus (CaMV) promoter (Odell et al., 1985) with the duplicated enhancer region (Kay et al., 1987). |
| cryIIIA | 1.8 | The gene which confers resistance to CPB. The gene encodes an amino acid sequence identical to the CPB control protein (referred to as the B.t.t. Band 3 protein) found in B.t.t. as described by Perlak et al. (1993). |
| E9 3' | 0.63 | A 3' nontranslated region of the pea ribulose-1,5-bisphosphate carboxylase small subunit (rbcS) E9 gene (Coruzzi et al., 1984), which functions to terminate transcription and direct polyadenylation of the cryIIIA mRNA. |
| NOS 3' | 0.26 | A 3' nontranslated region of the nopaline synthase gene which functions to terminate transcription and direct polyadenylation of the nptII mRNA (Depicker et al., 1982; Bevan et al., 1983). |
| nptII | 0.79 | The gene isolated from Tn5 (Beck et al., 1982) which encodes for neomycin phosphotransferase type II. Expression of this gene in plant cells confers resistance to kanamycin and serves as a selectable marker for transformation (Fraley et al., 1983). |
| 35S | 0.32 | The 35S promoter region of the cauliflower mosaic virus (CaMV) (Gardner et al., 1981; Sanders et al., 1987). |
| LB | 0.45 | A restriction fragment from the octopine Ti plasmid, pTi15955, containing the 24 bp T-DNA left border used to terminate the transfer of the T-DNA from Agrobacterium tumefaciens to the plant genome (Barker et al., 1983). |
| ori V | 1.3 | Origin of replication segment for ABI Agrobacterium derived from the broad-host range plasmid RK2 (Stalker et al., 1981). |
| ori-322/rop | 1.8 | A segment of pBR322 which provides the origin of replication for maintenance of the PV-STBT02 plasmid in E. coli, the replication of primer (rop) region and the bom site for the conjugational transfer into the Agrobacterium tumefaciens cells (Bolivar et al., 1977; Sutcliffe, 1978). |
| aad | 0.93 | A fragment isolated from transposon Tn7 containing a 0.79 kb gene which encodes for the enzyme streptomycin adenylyltransferase that allows for bacterial selection on spectinomycin or streptomycin (Fling et al., 1985). |
Table Notes
- Table Note 1
Sizes are approximations.
Figure 1: Example of a detailed map of a plasmid vector
(from APHIS petition #94-257-01p)
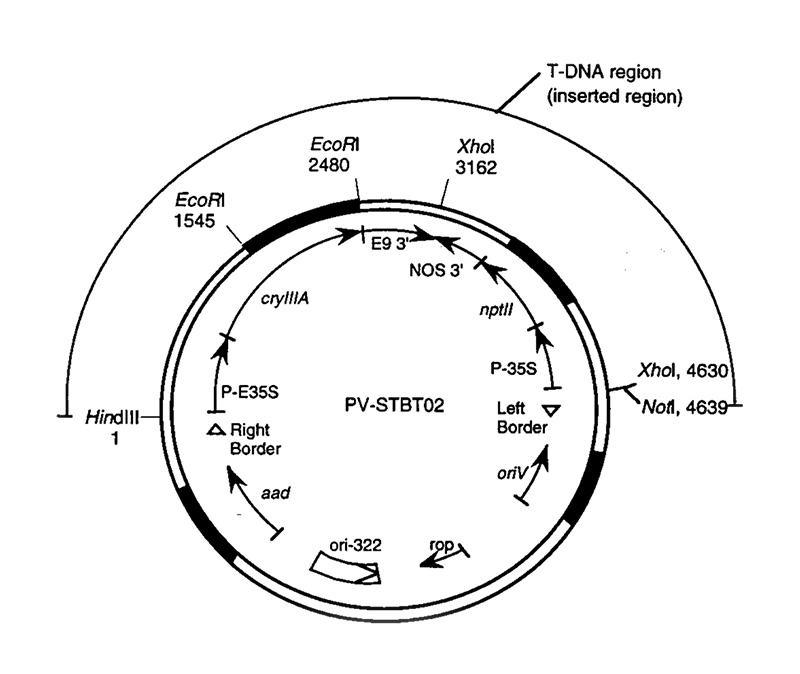
Description for images – Figure 1. Plasmid map of PV-STBT02.
Restriction sites, and their locations in base pairs, utilized during Southern analyses are shown. The region which served as the T-DNA is marked and it's delineating right and left borders are denoted by open arrows. The blackened regions denote positions of homology for PCR probes used during Southern analyses as described in Section V.A. Cleavage sites for HindIII, EcoRl, Xhot and Notl restriction endonucleases are shown. A description of the genetic elements appears in Table 1.
References
FAO/WHO (2000). Joint FAO/WHO Consultation on Foods Derived from Biotechnology: Safety aspects of genetically modified foods of plant origin. World Health Organization 29 May – 2 June 2000, WHO Headquarters, Geneva, Switzerland
FAO/WHO (2001). Joint FAO/WHO Consultation on Foods Derived from Biotechnology: Evaluation of the allergenicity of genetically modified foods, 22‑25 January 2001, Rome, Italy
OECD (1993). Safety evaluation of foods derived by modern biotechnology: concepts and principles. Organization for Economic Development and Cooperation, Paris.
- Date modified: